The CaDrA package currently provides four scoring functions to search for subsets of genomic features that are likely associated with a specific outcome of interest (e.g., protein expression, pathway activity, etc.)
- Kolmogorov-Smirnov Method (
ks) - Conditional Mutual Information Method (
revealer) - Wilcoxon Rank-Sum Method (
wilcox) - Custom - An User’s Provided Scoring Function
(
custom)
Below, we run candidate_search() over the top 3 starting
features using each of the four scoring functions described above.
Important Note:
- The legacy function
topn_eval()is equivalent to the recommendedcandidate_search()function
Load required datasets
- A
binary features matrixalso known asFeature Set(such as somatic mutations, copy number alterations, chromosomal translocations, etc.) The 1/0 row vectors indicate the presence/absence of ‘omics’ features in the samples. TheFeature Setmust be an object of class SummarizedExperiment from SummarizedExperiment package) - A vector of continuous scores (or
input_score) representing a functional response of interest (such as protein expression, pathway activity, etc.)
Kolmogorov-Smirnov scoring method
See ?ks_rowscore for more details
ks_topn_l <- CaDrA::candidate_search(
FS = sim_FS,
input_score = sim_Scores,
method = "ks_pval", # Use Kolmogorow-Smirnow scoring function
weight = NULL, # If weight is provided, perform a weighted-KS test
alternative = "less", # Use one-sided hypothesis testing
search_method = "both", # Apply both forward and backward search
top_N = 3, # Evaluate top 3 starting points for the search
max_size = 7, # Allow at most 7 features in meta-feature matrix
do_plot = FALSE, # We will plot it AFTER finding the best hits
best_score_only = FALSE # Return meta-feature, its observed input scores and corresponding best score
)
# Now we can fetch the feature set of top N features that corresponded to the best scores over the top N search
ks_topn_best_meta <- topn_best(ks_topn_l)
# Visualize best meta-feature result
meta_plot(topn_best_list = ks_topn_best_meta)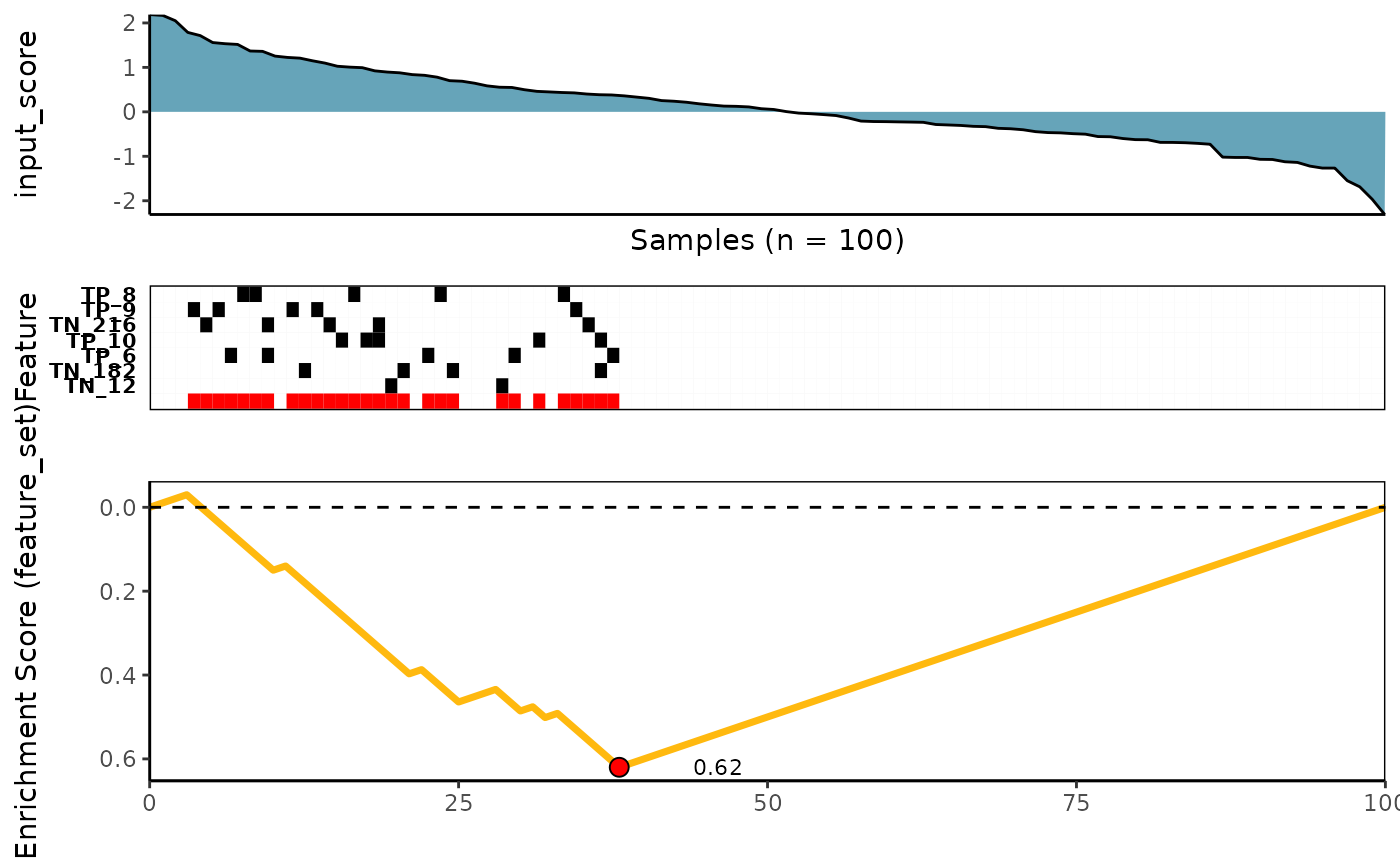
Wilcoxon Rank-Sum scoring method
See ?wilcox_rowscore for more details
wilcox_topn_l <- CaDrA::candidate_search(
FS = sim_FS,
input_score = sim_Scores,
method = "wilcox_pval", # Use Wilcoxon Rank-Sum scoring function
alternative = "less", # Use one-sided hypothesis testing
search_method = "both", # Apply both forward and backward search
top_N = 3, # Evaluate top 3 starting points for the search
max_size = 7, # Allow at most 7 features in meta-feature matrix
do_plot = FALSE, # We will plot it AFTER finding the best hits
best_score_only = FALSE # Return meta-feature, its observed input scores and corresponding best score
)
# Now we can fetch the feature set of top N feature that corresponded to the best scores over the top N search
wilcox_topn_best_meta <- topn_best(topn_list = wilcox_topn_l)
# Visualize best meta-feature result
meta_plot(topn_best_list = wilcox_topn_best_meta)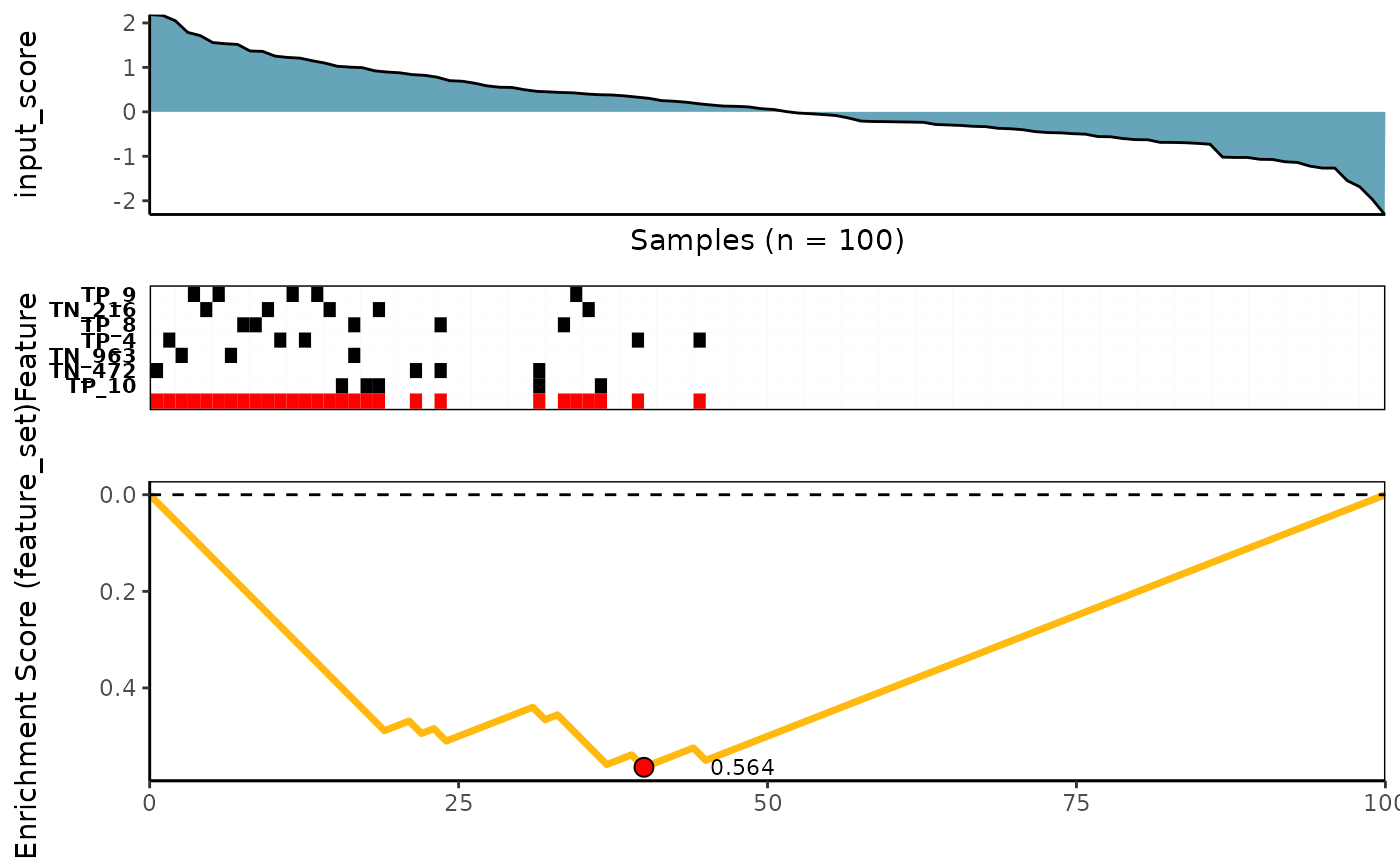
Conditional Mutual Information scoring method
See ?revealer_rowscore for more details
revealer_topn_l <- CaDrA::candidate_search(
FS = sim_FS,
input_score = sim_Scores,
method = "revealer", # Use REVEALER's CMI scoring function
search_method = "both", # Apply both forward and backward search
top_N = 3, # Evaluate top 3 starting points for the search
max_size = 7, # Allow at most 7 features in meta-feature matrix
do_plot = FALSE, # We will plot it AFTER finding the best hits
best_score_only = FALSE # Return meta-feature, its observed input scores and corresponding best score
)
# Now we can fetch the ESet of top feature that corresponded to the best scores over the top N search
revealer_topn_best_meta <- topn_best(topn_list = revealer_topn_l)
# Visualize best meta-feature result
meta_plot(topn_best_list = revealer_topn_best_meta)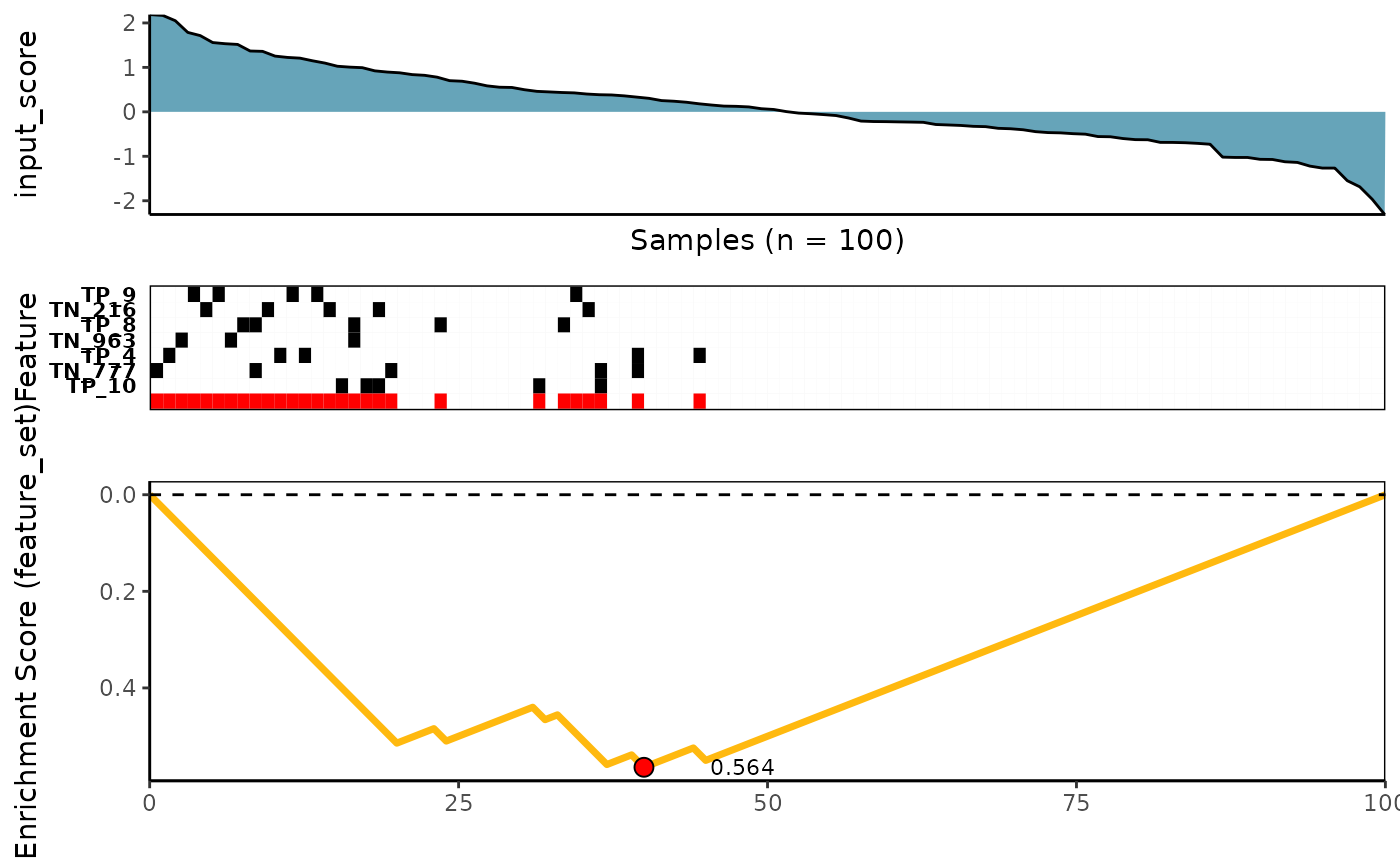
Custom - An user’s provided scoring method
See ?custom_rowscore for more details
# A customized function using ks-test function
customized_rowscore <- function(FS_mat, input_score, alternative){
ks <- apply(FS_mat, 1, function(r){
x = input_score[which(r==1)];
y = input_score[which(r==0)];
res <- ks.test(x, y, alternative=alternative)
return(c(res$statistic, res$p.value))
})
# Obtain score statistics and p-values from KS method
stat <- ks[1,]
pval <- ks[2,]
# Compute the -log scores for pval
scores <- -log(pval)
names(scores) <- rownames(FS_mat)
# Re-order FS in a decreasing order (from most to least significant)
# This comes in handy when doing the top-N evaluation of
# the top N 'best' features
scores <- scores[order(scores, decreasing=TRUE)]
return(scores)
}
# Search for best features using a custom-defined function
custom_topn_l <- CaDrA::candidate_search(
FS = sim_FS,
input_score = sim_Scores,
method = "custom", # Use custom scoring function
custom_function = customized_rowscore, # Use a customized scoring function
custom_parameters = list(alternative = "less"), # Additional parameters to pass to custom_function
search_method = "both", # Apply both forward and backward search
top_N = 3, # Evaluate top 3 starting points for the search
max_size = 7, # Allow at most 7 features in meta-feature matrix
do_plot = FALSE, # We will plot it AFTER finding the best hits
best_score_only = FALSE # Return meta-feature, its observed input scores and corresponding best score
)
# Now we can fetch the feature set of top N feature that corresponded to the best scores over the top N search
custom_topn_best_meta <- topn_best(topn_list = custom_topn_l)
# Visualize best meta-feature result
meta_plot(topn_best_list = custom_topn_best_meta)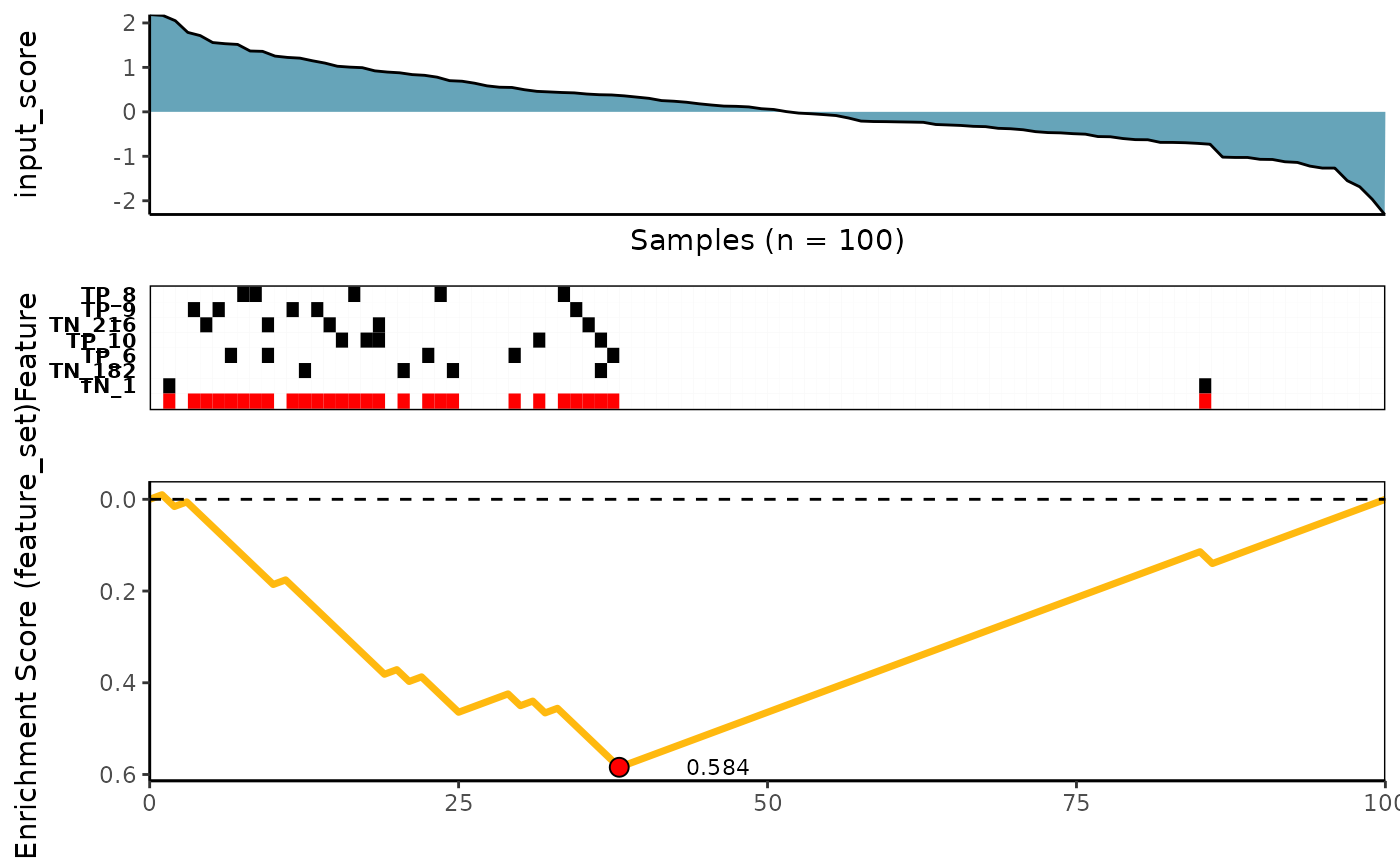
SessionInfo
sessionInfo()
R version 4.2.3 (2023-03-15)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 22.04.2 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.20.so
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] CaDrA_0.99.2 SummarizedExperiment_1.28.0
[3] Biobase_2.58.0 GenomicRanges_1.50.2
[5] GenomeInfoDb_1.34.9 IRanges_2.32.0
[7] S4Vectors_0.36.2 BiocGenerics_0.44.0
[9] MatrixGenerics_1.10.0 matrixStats_0.63.0
[11] BiocStyle_2.26.0
loaded via a namespace (and not attached):
[1] sass_0.4.5 jsonlite_1.8.4 foreach_1.5.2
[4] R.utils_2.12.2 gtools_3.9.4 bslib_0.4.2
[7] highr_0.10 BiocManager_1.30.20 GenomeInfoDbData_1.2.9
[10] yaml_2.3.7 pillar_1.8.1 lattice_0.20-45
[13] glue_1.6.2 digest_0.6.31 XVector_0.38.0
[16] colorspace_2.1-0 htmltools_0.5.4 Matrix_1.5-3
[19] R.oo_1.25.0 plyr_1.8.8 pkgconfig_2.0.3
[22] misc3d_0.9-1 bookdown_0.33 zlibbioc_1.44.0
[25] purrr_1.0.1 scales_1.2.1 tibble_3.2.0
[28] farver_2.1.1 ggplot2_3.4.1 withr_2.5.0
[31] cachem_1.0.7 ppcor_1.1 cli_3.6.0
[34] magrittr_2.0.3 memoise_2.0.1 evaluate_0.20
[37] R.methodsS3_1.8.2 fs_1.6.1 fansi_1.0.4
[40] R.cache_0.16.0 doParallel_1.0.17 MASS_7.3-58.2
[43] gplots_3.1.3 textshaping_0.3.6 tools_4.2.3
[46] lifecycle_1.0.3 stringr_1.5.0 munsell_0.5.0
[49] DelayedArray_0.24.0 compiler_4.2.3 pkgdown_2.0.7
[52] jquerylib_0.1.4 caTools_1.18.2 systemfonts_1.0.4
[55] rlang_1.1.0 grid_4.2.3 RCurl_1.98-1.10
[58] iterators_1.0.14 labeling_0.4.2 bitops_1.0-7
[61] tcltk_4.2.3 rmarkdown_2.20 gtable_0.3.1
[64] codetools_0.2-19 reshape2_1.4.4 R6_2.5.1
[67] knitr_1.42 fastmap_1.1.1 utf8_1.2.3
[70] rprojroot_2.0.3 ragg_1.2.5 KernSmooth_2.23-20
[73] desc_1.4.2 stringi_1.7.12 parallel_4.2.3
[76] Rcpp_1.0.10 vctrs_0.6.0 xfun_0.37